| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| 27 | 28 | 29 | 30 |
- 증권사레포트
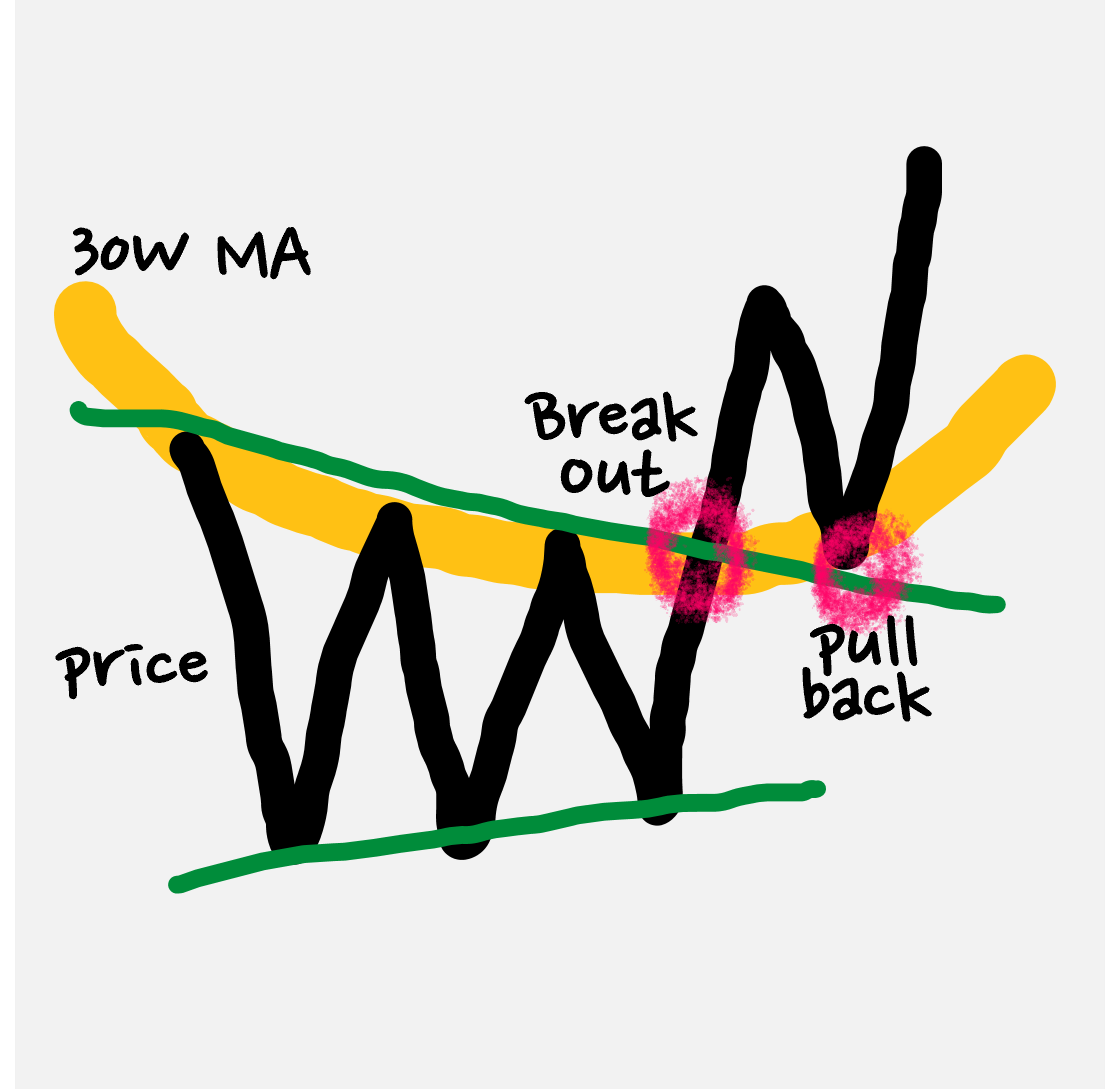
- 쌍바닥패턴
- 비교유전체
- 생명정보학
- 우분투
- bioinformatics
- 기본적분석
- 지지저항
- 주식매매
- 초단타
- 목표주가
- 리눅스
- 상한가
- 스캘핑
- 이동평균선
- 분봉차트
- 지지저항선
- mummer
- 매매일지
- 유전체
- 주식투자
- 차트분석
- 생물정보학
- 추천종목
- 매매기법
- 돌파매매
- W패턴
- 관심종목
- 기술적분석
- 세균
- Today
- Total
목록mummer (6)
A Fine-Tuned Universe
내가 사용한 show-coords 명령어 nucmer -p wt_lab wt.fasta lab.fasta show-coords -rcl wt_lab.delta > wt_lab.coords 이렇게 하면 테이블 형태로 결과가 정리된다
4: FINISHING DATA ERROR: Could not parse input from 'Query File'. Please check the filename and format, or file a bug report ERROR: postnuc returned non-zero 이 에러메시지가 왜 뜨는지 검색해보면 여러가지 의견이 많은데 내 경험상 query file을 우분투에서 저장하면 해결된다. 다시 말해서 윈도에서 GenBank 등에서 다운로드 받..
지난 번에 nucmer header 에 대해 정리했는데 이번엔 mummer 결과파일의 header에 정리해놓으려고 한다. 좋은 프로그램 같긴 한데 쓰려면 알아야 할 게 많은 것 같다. (결과 예시) > ID1 4655667 1 31 4655699 33 319 4656019 353 520 4656540 874 20 > ID1 Reverse 741743 22 872 (설명) > 뒤에는 시퀀스ID 가 나온다 (inpu..
nucmer --prefix=lab_high_nucmer lab.fasta high.fasta # Lab strain 과 high-resistance strain의 genome을 alignment show-snps -C -T lab_high_nucmer.delta > lab_high.snps SNP을 찾되 좀 더 확실한 mutation만 (이 부분은 잘 이해못했음) Tab-delimited 파일로 정리 다른 유전체 분석할 때는 fasta 파일 이름과 prefix 바꾸면 된다